![]()
![]()
![]()
hiPhive — High-order force constants for the masses#
hiPhive (pronounced /haɪ faɪv/) is a tool for efficiently extracting interatomic force constants (FCs) from atomistic simulations, most commonly density functional theory calculations, including higher-order FCs in large and/or low-symmetry systems. It is written in Python, which allows easy integration with countless first-principles codes and analysis tools accessible in Python, and allows for a simple and intuitive user interface. The FCs enable one to compute a variety of thermodynamic properties, including, e.g., phonon dispersions, free energies, thermal conductivities or defect lineshapes.
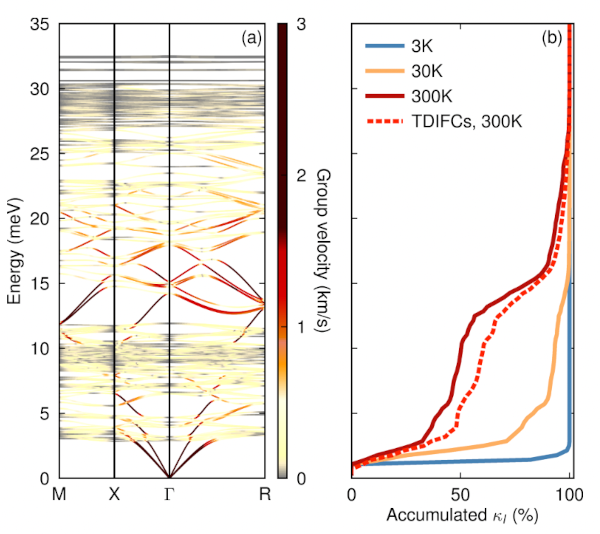
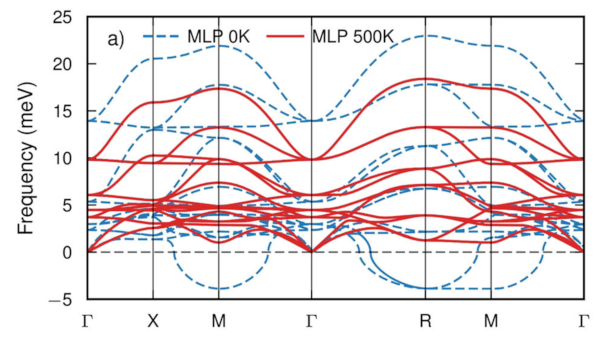
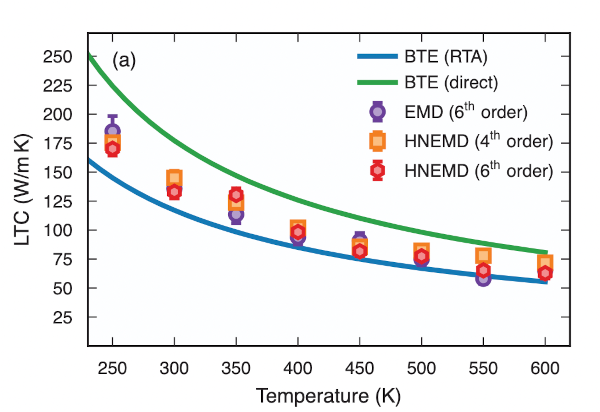
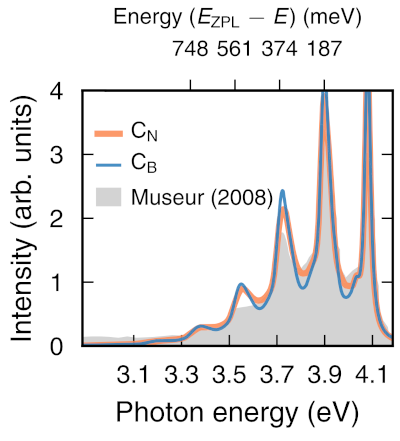
hiPhive does not rely on a specific type of input configuration (e.g., enumerated displacements or configurations from molecular dynamics simulations). Rather it employs advanced optimization techniques that are designed to find sparse solutions, which in the present case reflect the short-range nature of the FCs. If the input configurations are constructed sensibly this approach allows one to obtain FCs using a much smaller number of input configurations and thus to reduce the computational effort, usually in the form of density functional theory calculations, considerably. This approach becomes genuinely advantageous already for obtaining second-order FCs in large and/or low symmetry systems (defects, interfaces, surfaces, large unit cells etc). hiPhive truly excels when it comes to higher order FCs, for which a strict enumeration scheme quickly leads to an explosion of displacement calculations.
In addition to a comprehensive introduction provided in the get started section, a number of extensive example applications are available in the hiphive-examples repository.
The following snippet illustrates the construction of a force constant potential object (fcp), which can be subsequently used, e.g., for generating phonon dispersions, computing phonon lifetimes, or running molecular dynamics simulations:
cs = ClusterSpace(ideal_cell, cutoffs=[6.0, 4.5])
sc = StructureContainer(cs)
for atoms in list_of_training_structures:
sc.add_structure(atoms)
opt = Optimizer(sc.get_fit_data())
opt.train()
fcp = ForceConstantPotential(cs, opt.parameters)
hiPhive has been developed at the Department of Physics of Chalmers University of Technology (Gothenburg, Sweden) in the Condensed Matter and Materials Theory division. Please consult the credits page for information on how to cite hiphive.
For questions and help please use the hiphive discussion forum on matsci.org. hiPhive and its development are hosted on gitlab.